- Russian
- English
- Français
Мы на научных конференциях




Синдромы преждевременного старения
- Прогерии
- Синдром Вернера
- ДНК-геликазы
- Синдром Ротмунда-Томпсона (RTS)
- Синдром Блума
- Синдром Хатчинсона-Гилфорда
- Структура ядра
- Рестриктивная дермопатия
- Пигментная ксеродерма
- Трихотиодистрофия
- Синдром Кокейна
- Атаксия-телангиэктазия
- Синдром поломок Ниджмеджена (NBS)
- Анемия Фанкони
- Связь прогерий с теломерами
- Врожденный дискератоз
- Синдром Дауна
- Общие причины прогерий
- Перспективы исследований
- Исследовательские группы в области прогерий
- Ссылки на полные тексты статей по теме
- Похожие компасы
СИНДРОМЫ ПРЕЖДЕВРЕМЕННОГО СТАРЕНИЯ
Пожалуй наиболее ярким доказательством определяющей роли генов в старении являются моногенные болезни с признаками ускоренного старения (прогерии). О причинах данных заболеваний и их связи с естественным старением и пойдет речь.
Прогерии
Одним из подходов к изучению молекулярных основ старения человека является выяснение причин заболеваний преждевременного старения - так называемых частичных прогерий. В большинстве своем они моногенны, а значит, легко поддаются анализу. Недостатком данного подхода является то, что иногда их симптомы лишь напоминают свойства нормального старения, либо представлены не все свойства. Например, симптомы старения при прогериях более выражены и могут появляться в другой последовательности, чем в случае с "нормальным" старением. В частности, рост ногтей замедляется при старении, тогда как при прогериях с короткими теломерами останавливается полностью. Истончение бровей при старении следует за потерей волос на голове, но, наоборот, предшествует ему при прогериях.
Таким образом, мутации определенных генов у человека приводят к тяжелым заболеваниям, связанным с признаками преждевременного старения. Что это за заболевания и какие гены их обусловливают? Найдем ответы на эти вопросы.
Синдром Вернера
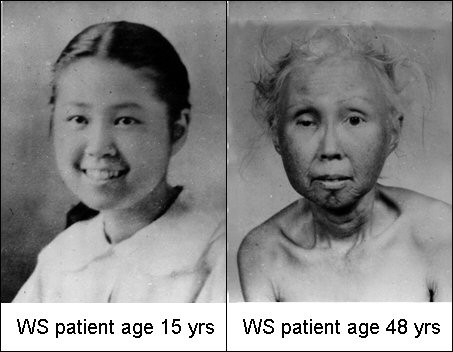
Одним из наиболее известных заболеваний с признаками ускоренного старения является синдром Вернера (http://en.wikipedia.org/wiki/Werner_syndrome, прогерия взрослых) - аутосомно-рецессивное заболевание (то есть контролируется рецессивными аллелями аутосомного гена), характеризующееся проявлением симптомов преждевременного старения кожи, сосудистой и репродуктивной системы, костей. До полового созревания пациенты развиваются нормально. Симптомы старения у них начинаются в ранней зрелости. Уже в молодом возрасте они страдают от катаракт, склеродермальных и дегенеративных сосудистых изменений, диабета и атеросклероза, остеопороза, высокой частоты некоторых видов рака, поседения. Больные преждевременно погибают либо от рака, либо от сердечно-сосудистой патологии. Средняя продолжительность жизни при данном заболевании - 40-50 лет.
Функция гена данного заболевания была охарактеризована первой среди генов всех прогерий (пионерская статья). При синдроме Вернера аутосомно-рецессивная мутация в гене WRN, находящемся на хромосоме 8, приводит к нарушению функции особой ДНК-геликазы. Основная роль белка WRN в клетке - реинициирование блокированных репликационных вилок. В результате мутации вызывается нарушение репликации и репарации ДНК, экспрессии генов, ускоренное укорочение теломер и повышенная чувствительность клеток к апоптозу (обзор).
ДНК-геликазы
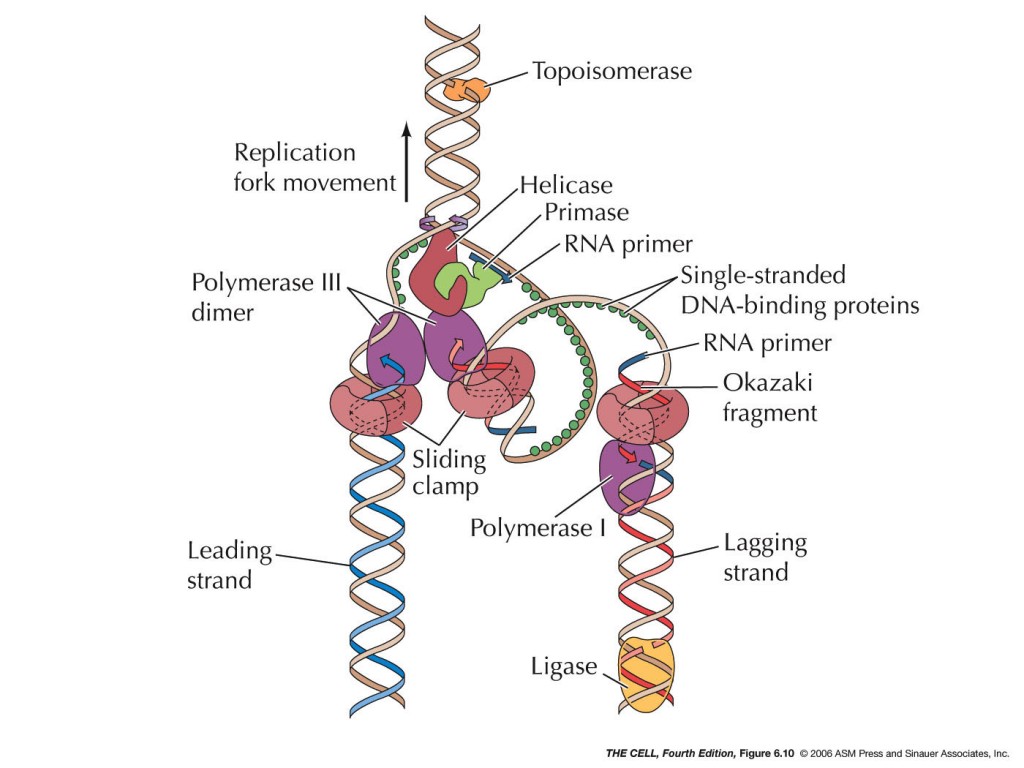
Здесь стоит напомнить, что такое ДНК-геликазы. Данные ферменты расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических процессов, таких как синтез копий ДНК (репликация), а также транскрипция РНК и репарация ДНК.
Синдром Ротмунда-Томпсона (RTS)

Для генетически близкого заболевания, аутосомно-рецессивного синдрома Ротмунда-Томпсона (http://en.wikipedia.org/wiki/Rothmund-Thompson's_syndrome), характерно наличие особой гиперпигментации кожи (пойкилодермия), гиперчувствительности к солнечным лучам, задержки роста, гипогонадотропного гипогонадизма, анемии, контрактуры мягких тканей, гиподонтии, ювенильных катаракт, проблем с ростом волос, остеогенной саркомы (последнее заболевание является отличительным признаком и при синдроме Вернера). Как и ген предыдущего заболевания, ген данного заболевания (RECQL4) относится к семейству RecQ 3'-5' ДНК геликаз, которые участвуют в поддержании стабильности генома через регуляцию репликационной вилки.
Синдром Блума

При аутосомно-рецессивном синдроме Блума (Bloom syndrome) отмечены гиперчувствительность к ультрафиолету, иммунодефицит, малорослость, остеосаркомы (являющиеся причиной смерти до 30 лет у больных с данным синдромом). Характерные для старения признаки менее выражены, чем при предыдущих синдромах, например, наблюдается преждевременная менопауза у женщин. Вследствие мутации в гене BLM, принадлежащем к генам ДНК-геликаз, синдром характеризуется нестабильностью генома и повышенным риском канцерогенеза.
Синдром Хатчинсона-Гилфорда
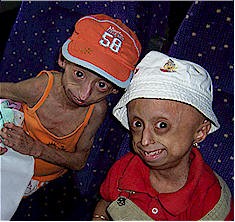
Очень часто, когда говорят о прогериях, имеют в виду именно синдром Хатчинсона-Гилфорда (Hutchinson-Gilford syndrome), так называемую "прогерию детей". Это крайне редкое заболевание (<1/1000000, тогда как частота предыдущих прогерий составляет в среднем < 1/100000). Еще одним отличием данной прогерии является то, что мутация, вызывающая ее, всегда возникает de novo, то есть не наследуется. Это не удивительно, поскольку носители погибают до репродуктивного возраста. Дети шестилетнего возраста при синдроме Хатчинсона-Гилфорда выглядят как уже пожилые люди и погибают от сильного атеросклероза к 13 годам. Данное заболевание отличают неспособность к росту, липоатрофия, костные нарушения, маленький клювообразный нос, срезанный подбородок, полная потеря волос, пятнистая гипопигментация кожи. С развитием заболевания возникают атеросклеротические бляшки, которые становятся проникающими, приводя к сердечным приступам и смерти.
Заболевание связано с дефектом гена структурного белка ядерной оболочки ламина А (lmna), что приводит к изменению структуры ядра, нестабильности генома и нарушению экспрессии генов. Мутация приводит к синтезу укороченной версии белка и, соответственно, к недостатку количества ламина А дикого типа.
Прогерия Хатчинсона-Джилфорда сопровождается дефектами в ядерной структуре и функциях: присутствует дизморфия поверхности ядра, увеличение уровня повреждений ДНК, снижение экспрессии ряда ядерных белков, включая важные гетерохроматиновые белки HP1 и LAP2 (из группы ламин А-ассоциированных белков). Кроме того, в клетках больных нарушен паттерн модифицированных гистонов: происходит снижение гетерохроматин-специфичного триметилирования по остатку Lys9 в гистоне H3 (статья). Таким образом, ядра клеток больных прогерией Хатчинсона-Джилфорда теряют гетерохроматин. В результате происходит патологическая сверхактивация ряда транскриптов, в норме репрессированных, например перицентрического сателлитного повтора III (статья). Коррекция сплайсинга ламина А в клетках пациентов восстанавливает: нормальную морфологию ядра, гетерохроматин-специфичную модификацию гистонов, экспрессию ряда дисрегулированых генов (статья).
Структура ядра
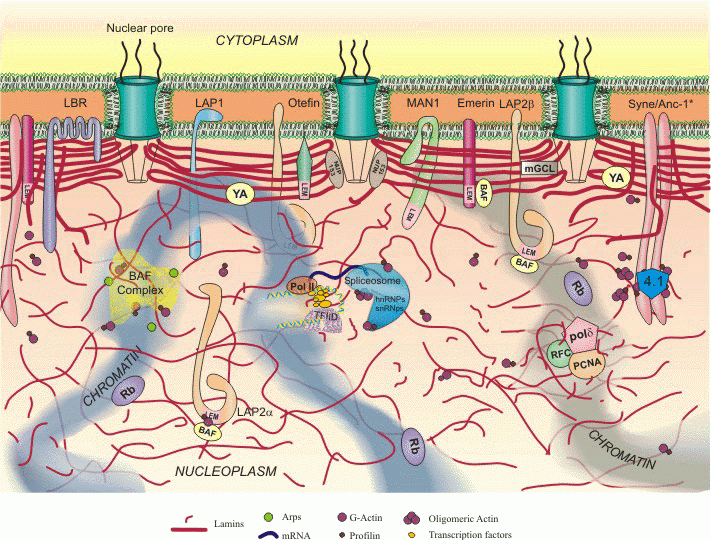
Таким образом, молекулярной причиной данного синдрома являются нарушения структуры ядра. Ядро клетки высших организмов является сложным, высокоорганизованным хранилищем индивидуальной генетической информации. Типичное ядро содержит особые функциональные области, представленные упорядоченно расположенными хромосомами и белковыми субкомпартментами, в которых происходят специфические процессы, включая экспрессию генов. В структурной организации ядра важную роль играет ядерная ламина. Она состоит из белков ламинов А и В типа. Эти промежуточные филаментные белки формируют переплетеную сеть, расположенную на периферии ядра и подстилающую ядерную мембрану. Ламина играет регуляторную роль в экспрессии генов, поскольку белки ламины взаимодействуют с хроматином и могут участвовать в фиксации и организации участков генома в пространстве. Ламина обеспечивает механические и поверхностные свойства ядра и является участком стыковки с периферическим гетерохроматином. Ламины распределены также в нуклеоплазме, где они участвуют в репликации ДНК и транскрипции с участием фермента РНК полимеразы II. Таким образом, нарушение ядерной ламины, взаимодействующей с хроматином, может приводить к нарушению экспрессии генов.
Рестриктивная дермопатия
Рестриктивная дермопатия (Restrictive dermopathy) представляет собой редкий аутосомно-рецессивный синдром. Это летальное перинатальное прогероидное заболевание, характеризуемое задержкой роста, плотной и жесткой кожей, облысением, микрогнатией и другими нарушениями костей, вызывается делецией гена ZMPSTE24, кодирующего протеазу, необходимую для эндопротеолитического процессинга преламина А в зрелый ламин. Таким образом, как и синдром Хатчинсона-Гилфорда, она вызывается дефектом биогенеза ламина А.
Пигментная ксеродерма

Есть целая группа генетических заболеваний, связанных с дефектами различных форм репарации ДНК и имеющих отдельные симптомы, указывающие на процессы ускоренного старения. Среди таких патологий одной из наиболее изученных является пигментная ксеродерма (Xeroderma pigmentosum) - редкое аутосомно-рецессивное заболевание, характеризуемое гиперчувствительностью к свету, ненормальной пигментацией и предрасположенностью к раку кожи, особенно на подверженных солнцу участках тела. Имеются различные симптомы от умеренных кожных нарушений до тяжелых повреждений кожи. Данное заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов. Существует семь комплементарных групп генов пигментной ксеродермы (от XPA до XPG). Группа G (XPG) кодирует белок с молекулярной массой 133 кДа, который имеет структура-специфичную эндонуклеазную активность и функционирует как 3'-нуклеаза в реакции двойного разрезания при эксцизионной репарации нуклеотидов. Первичные культуры эмбриональных фибробластов, полученные от XPG-дефицитных мышей, характеризуются активацией транскрипционного фактора р53 и преждевременным старением. Гены ксеродермы группы B и D (XPB, XPD) кодируют субъединицы транскрипционного фактора TFIIH.
Трихотиодистрофия

Заболевание трихотиодистрофия (Trichothiodystrophy) проявляется в пониженном содержании серы в волосах и в их повышенной ломкости, задержке умственного развития, гиперчувствительности к ультрафиолету и в других врожденных аномалиях. Причина заболевания - мутации в генах XPB и XPD, кодирующих субъединицы транскрипционного фактора TFIIH. Продукты генов XPB и XPD имеют геликазную и АТФ-азную активность и участвуют в формировании "транскрипционного пузырька", а также в эксцизионной репарации нуклеотидов, связанной с транскрипцией (Transcription-coupled repair).
Синдром Кокейна
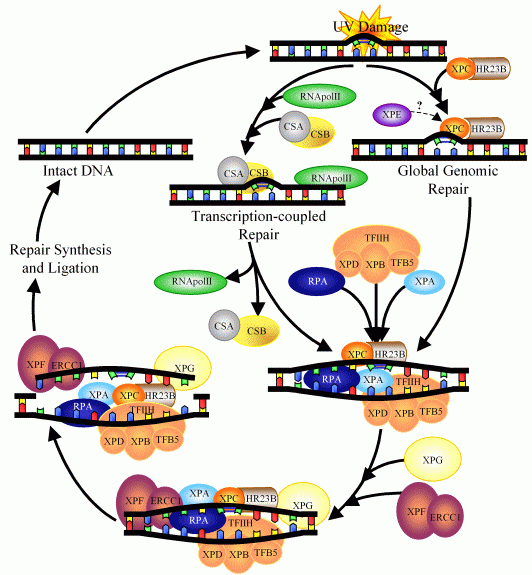
Синдром Кокейна (Cockayne syndrome) - редкое наследственное заболевание, при котором индивид не способен к росту, обладает короткой продолжительностью жизни и нейрологической дисфункцией. Клетки больных синдромом Кокейна чувствительны к ультрафиолету и окислительным формам повреждения ДНК, поскольку характеризуются дефектом одного из механизмов эксцизионной репарации нуклеотидов - связанного с транскрипцией репарационного процесса, предназначенного для удаления крупных повреждений транскрибируемых цепей ДНК активных генов. В разных вариантах синдрома Кокейна могут иметь место нарушения функции нескольких генов (CSA, CSB, XPD и XPG), каждый из которых вносит свой вклад во взаимодействие процессов репарации и транскипции, как показано на рисунке выше (статья).
Атаксия-телангиэктазия
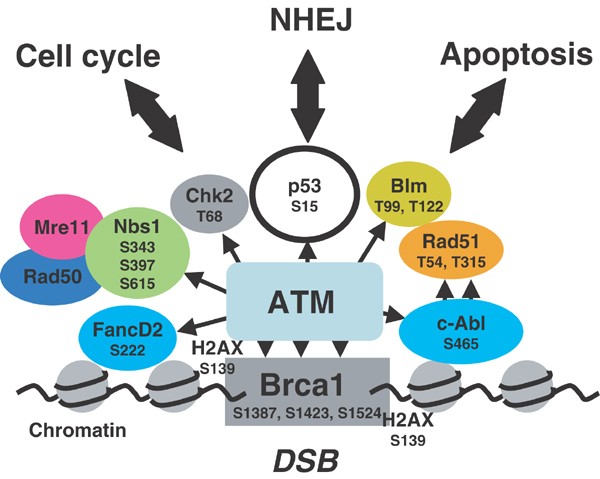
Больные аутосомно-рецессивным заболеванием атаксией-телангиэктазией (Ataxia telangiectasia) страдают от нейрональной дегенерации, преждевременного старения и увеличения частоты возникновения опухолей. In vitro клетки больных данным синдромом ускоренно теряют теломеры вследствие их оксидативного повреждения. Пациенты с атаксией-телангиэктазией несут мутацию в гене Atm (ataxia teleangiectasia mutated), кодирующем киназу ATM, главный сенсор повреждения ДНК в клетке. Распознавая повреждение ДНК в контрольных точках клеточного цикла, ATM фосфорилирует такие белки-мишени как р53, Chk1, Chk2, BRCA1, NBS1, FANCD2, гистон H2AX, которые, в свою очередь, индуцируют задержку клеточного цикла и репарацию ДНК. Мутации в генах некоторых мишеней ATM тоже приводят к ускоренному старению.
Синдром поломок Ниджмеджена (NBS)
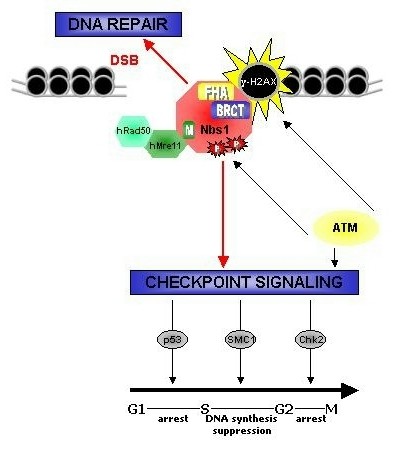
К таким мутациям относится и причина синдрома поломок Ниджмеджена (Nijmegen breakage syndrome). Больных отличает микроцефалия, особая форма лица, низкорослость, иммунодефицит, радиочувствительность, предрасположенность к лимфоидным разновидностям рака. В случае мутации гена NBS1 возникает нестабильность хромосом в результате дефекта структур Холлидея, образующихся в процессе пострепликативной рекомбинационной репарации двухцепочечных разрывов ДНК.
Анемия Фанкони

Симптомы анемии Фанкони (Fanconi anemia) - дефекты развития (например, отсутствие пальцев), нарушения функции красного костного мозга, острая миелогенная лейкемия и другие формы рака, зачастую не позволяющие дожить до зрелого возраста. Всего известно 7 генов, способных приводить к анемии Фанкони: FancA, FancB, FancC, FancD, FancE, FancF и FancG. Продукты этих генов фосфорилируются ATM и участвуют в репарации ДНК и задержке S-фазы клеточного цикла.
Связь прогерий с теломерами
Многие из перечисленных частичных прогерий сопряжены с короткими теломерами: синдром Вернера, RTS, прогерия Хатчинсона-Гилфорда, атаксия телангиэктазия, NBS. В свою очередь, ускоренное укорочение теломер при прогериях, обусловливая репликативное старение дифференцированных соматических клеток и дисфункцию стволовых клеток, вызывает симптомы, во многом напоминающие нормальное старение, такие как потеря волос, поседение, дистрофия ногтей, снижение костной массы, гематологические заболевания, иммунодефициты.
Врожденный дискератоз
Еще более убедительный пример роли теломер в прогериях и старении в целом - заболевание врожденный дискератоз (dyskeratosis congenita).
Врожденный дискератоз существует в двух формах. Х-сцепленная форма вызвана мутацией гена белка дискерина, участвующего в обеспечении функции теломеразы. Аутосомно-доминантная форма вызвана дефектом в гене РНК компонента теломеразы. В результате, пациенты с врожденным дискератозом имеют низкую активность теломеразы и короткие теломеры и умирают в ранней зрелости от нарушений в красном костном мозге, рака или легочных осложнений. Некоторые пациенты с врожденным дискератозом характеризуются высокой частотой карцином, что предполагает вклад укорочения теломер в возрастзависимое возникновение рака.
Синдром Дауна

Мало кто знает, но к синдромам ускоренного старения относится и синдром Дауна (Down_syndrome). Синдром Дауна вызывается трисомией по 21-й хромосоме и среди его фенотипов - ранее начало возрастзависимых патологических изменений, а также укорочение продолжительности жизни. В мозге больных обнаруживаются активированные ферменты каспаза-3 и каспаза-8, обусловливающие апоптоз нейронов в тех участках, где происходит накопление -амилоида и нейрофибриллярных бляшек (белок-предшественник амилоида может быть потенциальным субстратом каспаз). Культивируемые in vitro нейроны мышей с трисомией 16-й хромосомы (сходной с 21-й хромосомой человека) имеют меньшую продолжительность жизни, чем нормальные фетальные нейроны и их гибель предотвращается ингибиторами каспаз (статья).
Общие причины прогерий
Как мы видим, разнообразие частичных прогерий огромно. В то же время, четко выявляются механизмы, их обусловливающие:
- Нарушение свойств теломер, хроматина и клеточного ядра
- Нарушение репарации и репликации ДНК, генетическая нестабильность
- Нарушение экспрессии генов
- Репликативное старение
- Повышенная чувствительность клеток к апоптозу
- Элиминация стволовых клеток
Вполне вероятно, что те же самые механизмы задействованы в "нормальном" старении.
Перспективы исследований
Однако, следует признать, что не все еще ясно, например:
- В чем состоят сходства и в чем - различия частичных прогерий и обычного старения?
- Каким образом связаны между собой генетические причины всех прогерий, что является определяющим фактором?
- Какие из симптомов прогерий являются причиной, а какие - следствием ускоренного старения?
- Как противостоять ускоренному старению?
Исследовательские группы в области прогерий

Группа Паолы Скаффиди активно занимается исследованием молекулярных причин прогерии Хатчинсона-Гилфорда.
Сайт граппы:
http://ccr.cancer.gov/staff/staff.asp?profileid=6671
Фонд Мило Гладстейн по изучению синдрома Блума
Сайт фонда:
www.milogladsteinfoundation.org
Общество по изучению пигментной ксеродермы (XP Society)
Сайт общества:

Группа Лерри Леба, изучающая молекулярную генетику синдрома Вернера.
Сайт группы:
http://depts.washington.edu/loeblabs/research/research.htm
Ссылки на полные тексты статей по теме
- Молекулярные основы прогерий
- Гены, ответственные за прогерии у человека
- Синдромы преждевременного старения: проникновение вглубь процессов старения
- Новые подходы к прогериям
- Синдром прогерии Хатчинсона-Гилфорда
- Генетические детерминанты здорового долголетия и продолжительности жизни человек...
- WRN на теломерах: вклад в старение и рак
- Синдром Вернера и Функция белка Вернера
- Механизмы сердечнососудистых заболеваний при синдромах ускоренного старения